


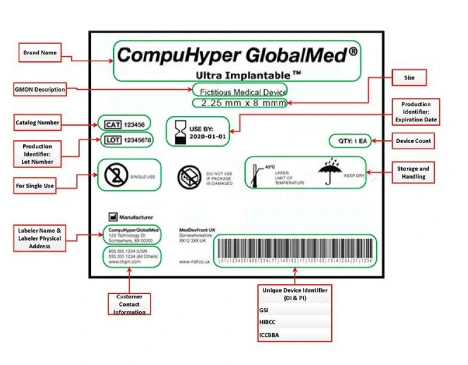

GUDID Account

UDI Submission

Time Required

30-Min Expert Consultation

Discount on Multiple UDIs



GUDID Account
UDI Submission
Time Required
30-Min Expert Consultation
Discount on Multiple UDIs